Overview of the 2023 ESC guidelines for the management of cardiomyopathies
EDITORIALS
Overview of the 2023 ESC guidelines for the management of cardiomyopathies
Article Summary
- DOI: 10.24969/hvt.2023.440
- Page(s): 264-268
- CARDIOVASCULAR DISEASES
- Published: 03/12/2023
- Received: 24/11/2023
- Accepted: 25/11/2023
- Views: 39505
- Downloads: 8609
- Keywords: guidelines, cardiomyopathy, hypertrophic cardiomyopathy, dilated cardiomyopathy, non-dilated left ventricular cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, restrictive cardiomyopathy, management
Address for Correspondence*: Yuriy Ivaniv, Danylo Halytsky National Medical University, Lviv, Ukraine
Email: yivaniv@gmail.com
Yuriy Ivaniv ORCID: 0000-0002-2153-9262 Facebook - Ivaniv Yuriy
Nelya Oryshchyn ORCID: 0000-0003-3758-9181 Facebook Oryshchyn Nelya
Yuriy Ivaniv, Nelya Oryshchyn, Anastasiya Aker
Danylo Halytsky National Medical University, Lviv, Ukraine
Graphical abstract
Key words: guidelines, cardiomyopathy, hypertrophic cardiomyopathy, dilated cardiomyopathy, non-dilated left ventricular cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy, restrictive cardiomyopathy, management
The objective of 2023 ESC Guideline (1) is to help healthcare professionals to diagnose and manage patients with cardiomyopathies (CMP ) according to the best clinical evidence. There are few randomized controlled clinical trials in patients with CMP, so the majority of the recommendations in this guidelines are based on observational cohort studies and expert consensus documents. Most of the recommendations in these guidelines are new, with the exception of the section on hypertrophic cardiomyopathy (HCM), in which the updated 2014 ESC guidelines on diagnosis and management of HCM are provided. The guidelines recommend an approach to disease diagnosis that is based on the predominant cardiac phenotype at presentation.
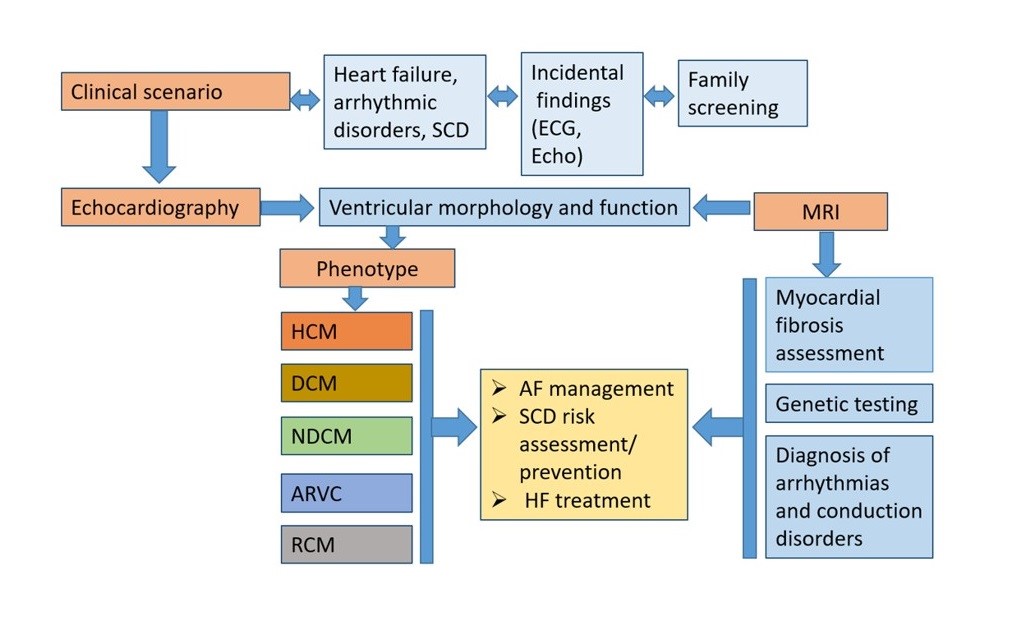
The diagnostic workflow of cardiomyopathy begins from the clinical scenario, which could be based on clinical symptoms, incidental findings or family screening following the diagnosis in relatives. Patients with CMP usually manifest with symptoms of heart failure or arrhythmic disorders.
The morphological and functional characterization in CMP is essential using a multiparametric approach. Morphological traits of CMP include ventricular (left or right) hypertrophy and/or ventricular dilatation.
Functional characterization includes ventricular systolic dysfunction (global or regional) and/or diastolic dysfunction. For the first time the guidelines underlie that the presence of non-ischaemic ventricular scar or fatty replacement on cardiac magnetic resonance imaging (CMR) and/or pathological examination, which can occur with or without ventricular dilatation and/or systolic dysfunction, can be the sole clue to the diagnosis of a cardiomyopathy. The systemic approach starting from the phenotype at presentation including tissue characterization enables clinicians to establish precise phenotype-based integrated diagnosis.
The guidelines propose classification with identification of 5 phenotypes of cardiomyopathies: dilated CMP, hypertrophic CMP, arrhythmogenic right ventricular CMP, non-dilated CMP and restrictive CMP. This nomenclature prompts clinicians to consider cardiomyopathy as the cause of several clinical presentations (e.g. arrhythmia, heart failure), and focuses on morphological and functional characteristics of the myocardium.
Hypertrophic cardiomyopathy is defined as the presence of increased LV wall thickness (with or without right ventricular (RV) hypertrophy) or left ventricular (LV) mass that is not solely explained by abnormal loading conditions.
Dilated cardiomyopathy (DCM) is defined as the presence of LV dilatation and global or regional systolic dysfunction unexplained solely by abnormal loading conditions (e.g. hypertension, valve disease, congenital heart disease) or coronary artery disease. Right ventricular dilatation and dysfunction may be present but are not necessary for the diagnosis of DCM.
For the first time new phenotype of the non-dilated left ventricular CMP (NDLVC) phenotype was presented. It is defined in the present guidelines as the presence of non-ischaemic LV scarring or fatty wall replacement regardless of the presence of global or regional wall motion abnormalities (RWMAs), or isolated global LV hypokinesia without scarring. The NDLVC phenotype will include individuals that up until now may have variably been described as having DCM (but without LV dilatation), arrhythmogenic left ventricular cardiomyopathy (ALVC), left dominant ARVC, or arrhythmogenic DCM (but often without fulfilling diagnostic criteria for ARVC). The identification of an NDLVC phenotype should trigger a multiparametric approach that leads to a specific etiological diagnosis, with implications for clinical treatment.
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is defined as the presence of predominantly RV dilatation and/or dysfunction concomitantly with histological involvement and/or electrocardiographic abnormalities in accordance with published criteria.
Restrictive cardiomyopathy (RCM) is defined as restrictive left and/or RV pathophysiology in the presence of normal or reduced diastolic volumes (of one or both ventricles), normal or reduced systolic volumes, and normal ventricular wall thickness.
The most important changes in this guideline relate to the group of conditions variously included under the umbrella term ‘arrhythmogenic cardiomyopathies’ (ACM). This term refers to a group of conditions that feature structural and functional abnormalities of the myocardium (identified by cardiac imaging and/or macroscopic and microscopic pathological investigation) and ventricular arrhythmia. A lack of a generally accepted definition has meant that the term encompasses a broad range of diverse pathologies and has introduced a number of inconsistencies and contradictions when applied in a clinical setting. The Task Force agreed to highlight the vital importance of arrhythmia as a diagnostic red flag and prognostic marker across a range of clinical phenotypes, but did not recommend the use of the term ACM as a distinct cardiomyopathy subtype as it lacks a morphological or functional definition consistent with the existing classification scheme.
The Task Force does not consider left ventricular non-compaction to be a CMP in the general sense. Instead, it is seen as a phenotypic trait that can occur either in isolation or in association with other developmental abnormalities, ventricular hypertrophy, dilatation, and/or systolic dysfunction. Given the lack of morphometric evidence for ventricular compaction in humans, the term ‘hypertrabeculation’, rather than LVNC, is recommended, particularly when the phenomenon is transient or clearly of adult onset.
Takotsubo syndrome is sometimes referred to as takotsubo or stress cardiomyopathy. Given the transient nature of the phenomenon, the Task Force does not recommend its classification as a cardiomyopathy.
The current Guidelines underline, that a multidisciplinary approach to patient care and appropriate transition of care from paediatric to adult CMP services is needed. It is recommended that all patients with cardiomyopathy and their relatives should have access to multidisciplinary teams with expertise in the diagnosis and management of CMPs. Timely and adequate preparation for transition of care from pediatric to adult services, including joint consultations, is recommended in all adolescents with CMP.
A multiparametric approach to the evaluation of patients with suspected cardiomyopathy is recommended, with the aims of
- establishing and characterizing the presence of a cardiomyopathy phenotype;
- identifying the underlying etiological diagnosis.
It is recommended that all patients with suspected or established CMP undergo systematic evaluation using a multiparametric approach that includes clinical evaluation, pedigree analysis, electrocardiography (ECG), Holter monitoring, laboratory tests, and multimodality imaging.
Patients with CMP may experience dyspnea, chest pain, palpitation, and syncope/or pre-syncope, although many individuals complain of few, if any, symptoms. A number of noncardiac symptoms (mental retardation, hear loss, learning difficulties, peripheral muscle weakness, acroparesthesia, deafness, skin abnormalities) act as pointers (or so called “red flags”) for specific etiology of cardiomyopathy phenotype. Similarly, general physical examination can provide diagnostic clues in patients with syndromic or metabolic causes of CMP.
Although the ECG is often non-specific, there are particular features that can suggest a certain etiology or morphological diagnosis, including atrioventricular (AV) block, ventricular pre-excitation pattern, distribution of repolarization abnormalities, and extremely high or low QRS voltages.
Routine (first-level) laboratory tests are recommended in all patients with suspected or confirmed CMP to evaluate etiology, assess disease severity, and aid in detection of extracardiac manifestations and assessment of secondary organ dysfunction. Additional (second-level) tests should be considered in patients with CMP and extracardiac features to aid in detection of metabolic and syndromic causes, following specialist evaluation.
Clinicians should approach a patient with suspected CMP using a ‘cardiomyopathy mindset’: use multimodality imaging to characterize the phenotype and identify abnormal ventricular morphology (e.g. hypertrophy, dilatation) and function (systolic/diastolic, global/regional), and detect abnormalities of tissue characterization (e.g. non-ischaemic myocardial scar and fatty replacement).
A comprehensive echocardiographic evaluation of cardiac dimensions and LV and RV systolic (global and regional) and LV diastolic function is recommended in all patients with CMP at initial evaluation, and during follow-up, to monitor disease progression and aid risk stratification and management. Transthoracic echocardiography provides relevant information on global and regional RV and LV anatomy and function as well as valve function and the presence of dynamic obstruction, pulmonary hypertension, or pericardial effusion.
Multimodality imaging is necessary to characterize the cardiac phenotype (morphology and function) – including tissue characterization for non-ischaemic myocardial scar detection, in combination with a detailed personal and family history, clinical examination, ECG and laboratory investigations. Tissue characterization by CMR is of value in diagnosis, disease progression monitoring and risk stratification in each of the main cardiomyopathy phenotypes.
Contrast-enhanced CMR is recommended in patients with CMP at initial evaluation (Class I Level B). Contrast-enhanced CMR should be considered in patients with cardiomyopathy during follow-up to monitor disease progression and aid in risk stratification and management (Class IIa Level C). Contrast-enhanced cardiac computed tomography (CT) should be considered in patients with suspected cardiomyopathy who have inadequate echocardiographic imaging and contraindications to CMR. Contrast-enhanced CMR should be considered for the serial follow-up and assessment of therapeutic response in patients with cardiac amyloidosis, Anderson–Fabry disease, sarcoidosis, inflammatory cardiomyopathies, and haemochromatosis with cardiac involvement (Class IIa Level B). Contrast-enhanced CMR should be considered in genotype-positive/phenotype-negative family members to aid in diagnosis and detect early disease in families with cardiomyopathy in which a disease-causing variant has been identified (Class IIa Level B).
In patients with suspected cardiomyopathy, endomyocardial biopsy should be considered to aid in diagnosis and management when the results of other clinical investigations suggest myocardial inflammation, infiltration, or storage that cannot be identified by other means (Class IIa Level C).
Genetic testing should be performed in patients with CMP, and may influence risk stratification and management. Genetic counselling, including pre- and post-test counselling, and psychological support are an essential aspect of the multidisciplinary care of patients with CMP and their relatives.
It is recommended that all patients with suspected CMP should undergo evaluation of family history and that a three- to four-generation family tree is created to aid in diagnosis, provide clues to underlying etiology, determine inheritance pattern, and identify at-risk relatives.
Genetic testing is recommended for patients who fulfill diagnostic criteria for CMP in cases where it enables diagnosis, prognostication, therapeutic stratification, or reproductive management of the patient, or where it enables a cascade genetic evaluation of the patient’s relatives who would otherwise be enrolled into long-term surveillance (Class I Level B).
Genetic testing in individuals with CMP, often referred to as confirmatory or diagnostic testing, serves the primary purpose of confirming the diagnosis. Additionally, it can provide insights into prognosis, aid in the selection of appropriate treatments, guide reproductive decisions, offer benefits to family members, and provide essential psychological support to patients struggling to understand their disease. Genetic testing of an affected individual may be indicated, even if it is unlikely to alter their management, if there are relatives who may benefit from testing. Postmortem genetic testing is recommended for a deceased individual who had CMP if a genetic diagnosis would facilitate the management of surviving relatives.
Symptom management, identification and prevention of disease-related complications (including sudden cardiac death (SCD), heart failure, and stroke) are the cornerstone of management of all cardiomyopathies.
Sudden cardiac death risk: Ventricular arrhythmias, particularly in the form of electrical storm and/ or repetitive appropriate Implantable cardioverter defibrillator (ICD) interventions, contribute to a significantly increased risk of morbidity and mortality in patients with cardiomyopathies. ICDs are effective at correcting potentially lethal ventricular arrhythmias and preventing SCD, but are also associated with complications, particularly in young patients who will require several replacements during their lifetimes. ICDs reduce mortality in survivors of cardiac arrest and in patients who have experienced haemodynamically compromising sustained ventricular arrhythmias, thus implantation of an ICD is recommended in patients with HCM, DCM, and ARVC who have survived a cardiac arrest due to ventricular tachycardia or ventricular fibrillation (Class I Level B), or who have spontaneous sustained ventricular arrhythmia causing syncope or haemodynamic compromise in the absence of reversible causes (Class I Level C).
Primary SCD prevention: Comprehensive SCD risk stratification is recommended in all CMP patients who have not suffered a previous cardiac arrest/sustained ventricular arrhythmia at initial evaluation and at 1–2 year intervals, or whenever there is a change in clinical status. The use of validated SCD algorithms/scores as aids to the shared decision-making when offering ICD implantation, where available: is recommended in patients with HCM (Class I Level B), should be considered in patients with DCM, NDLVC, and ARVC (Class IIa Level B).
Validated SCD risk-prediction tools (HCM Risk-SCD and HCM Risk-Kids) are the first step in sudden death prevention in patients with HCM. Additional risk markers may be of use in patients with low or intermediate risk. Implantation of an ICD should be considered in patients with HCM with an estimated 5-year risk of sudden death of ≥6%, following detailed clinical assessment (Class IIa, Level B). For patients with HCM who are in the low-risk category (<4% estimated 5-year risk of SCD), the presence of extensive late gadolinium enhancement (≥15%) on CMR and the presence of LV ejection fraction <50% may be considered in shared decision-making with patients about prophylactic ICD implantation (Class IIb, Level B).
An ICD implantation should be considered to reduce the risk of sudden death and all-cause mortality in patients with DCM, symptomatic heart failure, and LV ejection fraction ≤35% despite >3 months of optimal medical therapy (Class IIa, Level A). An ICD should be considered in patients with DCM with a genotype associated with high SCD risk and LVEF >35% in the presence of additional risk factors (Class IIa, Level C).
The clinical management of heart failure has been described in the 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure (2). In that document, recommendations are generally independent from the aetiology of heart failure and include current medical therapy, devices, and LV assist device (LVAD)/transplantation.
Pharmacological treatment of DCM patients does not differ from those recommended in chronic heart failure.
Patients with HCM and heart failure should receive guideline-directed medical therapy (GDMT) tailored to the specific phenotype of heart failure due to ejection fraction (as usual heart failure with preserved ejection fraction (HFpEF)). In the end-stage HCM treatment of heart failure with reduced ejection fraction (HFrEF) should be used. In patients with HCM and atrial fibrillation a rhythm control strategy is preferred, especially before significant left atrial remodeling/dilatation occurs. HCM patients could have a high atrial fibrillation recurrences rate, but catheter ablation in up to two-thirds of HCM patients may achieve maintenance of sinus rhythm based on data from several registers.
Oral anticoagulants are indicated for all patients with HCM and atrial fibrillation (unless contraindicated), regardless of the CHA2DS2-Vasc2 score (Class IB).
It is recommended to consider LV outflow tract obstruction (LVOTO) treatment in symptomatic HCM patients with a peak outflow tract gradient ≥50 mm Hg. Beta-blockers are the first-line medical therapy with class I indication, verapamil or diltiazem can be used as the second-line therapy with cautious titration from a small dose. Furthermore, mavacamten, a novel pharmacological agent that is a cardiac myosin ATPase inhibitor, should be considered as a treatment option for patients who remain symptomatic despite beta-blocker therapy. If severe symptoms or unexplained recurrent syncope persist, despite medical therapy, invasive septal reduction therapy is recommended. It is underlined, that septal reduction therapy should be performed by experienced multidisciplinary team expert in the management of HCM.
It is of importance to define etiology for a tailored management in patients with syndromic and metabolic cardiomyopathies (i.e. ERT/chaperone in lysosomal storage disease; tafamidis in ATTRwt, etc.). Cardiac amyloidosis and some forms of RCM deserve special consideration regarding heart failure management. Fluid control and maintenance of euvolaemia are central. If heart failure symptoms are present, loop diuretics should be given, although orthostatic hypotension may cause intolerance, and excessive fluid loss may worsen symptoms due to restriction (e.g. in HCM or amyloidosis).
In patients with end-stage heart failure and cardiomyopathy, regardless of the phenotype, orthotopic cardiac transplantation is recommended if they meet established eligibility criteria (Class IC). Also, it can include individuals with RCM and HCM who have normal LV ejection fraction and severe drug-refractory symptoms. Additionally, refractory ventricular tachycardia can serve as an indication for considering cardiac transplantation.
Exercise recommendations for patients with cardiomyopathy. Due to different phenotypes of CMPs or genetic screening, it is crucial to take into account special clinical and diagnostic scenarios. Regular low- to moderate-intensity exercise is recommended in all able individuals with cardiomyopathy. An individualized risk assessment for exercise prescription is recommended in all patients with cardiomyopathy. Evaluation should be guided by three principles: preventing life-threatening arrhythmias during exercise; symptom management to allow sports; and preventing sports-induced progression of the arrhythmogenic condition. Individuals who are genotype-positive/phenotype-negative or have a mild CMP phenotype and absence of symptoms or any risk factors, may be able to participate in competitive sports. In some high-risk patients with HCM, ARVC, DCM and NDLVC, high-intensity exercise and competitive sports should be discouraged.
The Guidelines recommend a multidisciplinary approach to patient care, and underline the need for integrated care between members of CMP teams, including cardiologists, radiologists, geneticists, cardiac surgeons, electrophysiologists, pathologists, psychologists, and other specialties.
Peer-review: Internal
Conflict of interest: None to declare
Authorship: Y.I, N.O. and A.A. equally contributed to preparation of manuscript
Acknowledgment and Funding: None to declare
References
|
||||||||||||||||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
