Unraveling the association between oxidative stress, nitric oxide signaling, and hypoxia inducible factor in chronic limb-threatening ischemia
REVIEW
Unraveling the association between oxidative stress, nitric oxide signaling, and hypoxia inducible factor in chronic limb-threatening ischemia
Article Summary
- DOI: 10.24969/hvt.2026.633
- CARDIOVASCULAR DISEASES
- Published: 10/03/2026
- Received: 04/02/2026
- Revised: 28/02/2026
- Accepted: 28/02/2026
- Views: 1130
- Downloads: 915
- Keywords: Oxidative stress, chronic limb-threatening ischemia, endothelial dysfunction, nitric oxide, hypoxia inducible factor
Address for Correspondence: Mohammed H. Hassan, Department of Medical Biochemistry and Molecular Biology, Faculty of Medicine, Qena University, Qena, 83523, Egypt; Department of Pathology, College of Medicine, Qassim University, Buradayh, Kingdom of Saudi Arabia
E-mail: mohammedhosnyhassaan@yahoo.com; mohammedhosnyhassaan@med.svu.edu.eg Mobile +201098473605
ORCID: Mohammed H. Hassan-0000-0003-2698-9438; Mohamed O. Mahmoud- 0000-0002-6737-8142;
Maha M. Mahmoud-0009-0007-2776-8668; Zeinab Mohammed Askary-0000-0003-3831-7911;
Mohamed A. Tohamy-0000-0002-5232-4766
Mohammed H. Hassan1a,2*, Mohamed O. Mahmoud3, Maha M. Mahmoud3, Zeinab Mohammed Askary1b, Mohamed A. Tohamy3
1aDepartment of Medical Biochemistry and Molecular Biology, 1bDepartment of Vascular Surgery, Faculty of Medicine, Qena University, Qena, 83523, Egypt
2Department of Pathology, College of Medicine, Qassim University, Buradayh, Kingdom of Saudi Arabia
3Department of Biochemistry, Faculty of Pharmacy, Beni-Suef University, Beni-Suef, 62514, Egypt
Abstract
Objective: Chronic limb-threatening ischemia (CLTI) is a serious medical condition characterized by decreased blood supply to the lower limbs, which causes tissue loss and amputation. The pathophysiology of CLTI has been associated with oxidative stress, nitric oxide (NO) imbalance, and the hypoxia-inducible factor (HIF) signaling pathway. This review focuses on how oxidative stress causes endothelial dysfunction, impairment of NO bioavailability, and HIF stability, subsequently aggravating tissue hypoxia and ischemia. Furthermore, this review explores the treatment implications of focusing on these pathways to enhance CLTI outcomes. The aim of the review was to explore the role of oxidative stress, NO imbalance, and HIF pathway dysregulation in CLTI pathophysiology and implications for treatment.
Methods: PubMed, Google Scholar, E-library, and the websites of medical journals (Medicine, Biology, and Biochemistry) were used as electronic databases for the literature search. Overall, 109 articles out of the 214 that were initially discovered satisfied the inclusion requirements.
Results: Oxidative stress contributes to CLTI progression via endothelial dysfunction, impaired nitric oxide bioavailability, and HIF stability, worsening tissue hypoxia and ischemia.
Conclusion: Targeting oxidative stress, nitric oxide, and HIF pathways may enhance CLTI outcomes.
Key words: Oxidative stress, chronic limb-threatening ischemia, endothelial dysfunction, nitric oxide, hypoxia inducible factor
List of abbreviations
ABPI Ankle-Brachial Pressure Index
AGEs Advanced Glycation End Products
BH4 Tetrahydrobiopterin
cGMP Cyclic Guanosine Monophosphate
CLTI Chronic Limb-Threatening Ischemia
EC Endothelial Cell
GSH Glutathione
HDLs High-Density Lipoprotein
HIF Hypoxia Inducible Factor
NO Nitric Oxide
NOS Nitric Oxide Synthase
NOXs NADPH Oxidases
oxLDL Oxidized Low-Density Lipoprotein
PAD Peripheral Arterial Disease
PHD Prolyl Hydroxylases
PKC Protein Kinase C
ROS Reactive Oxygen Species
SNP Single-Nucleotide Polymorphisms
TBI Toe-Brachial Index
VEGF Vascular Endothelial Growth Factor
VNTR Variable Numbers of Tandem Repeats

Graphical abstract
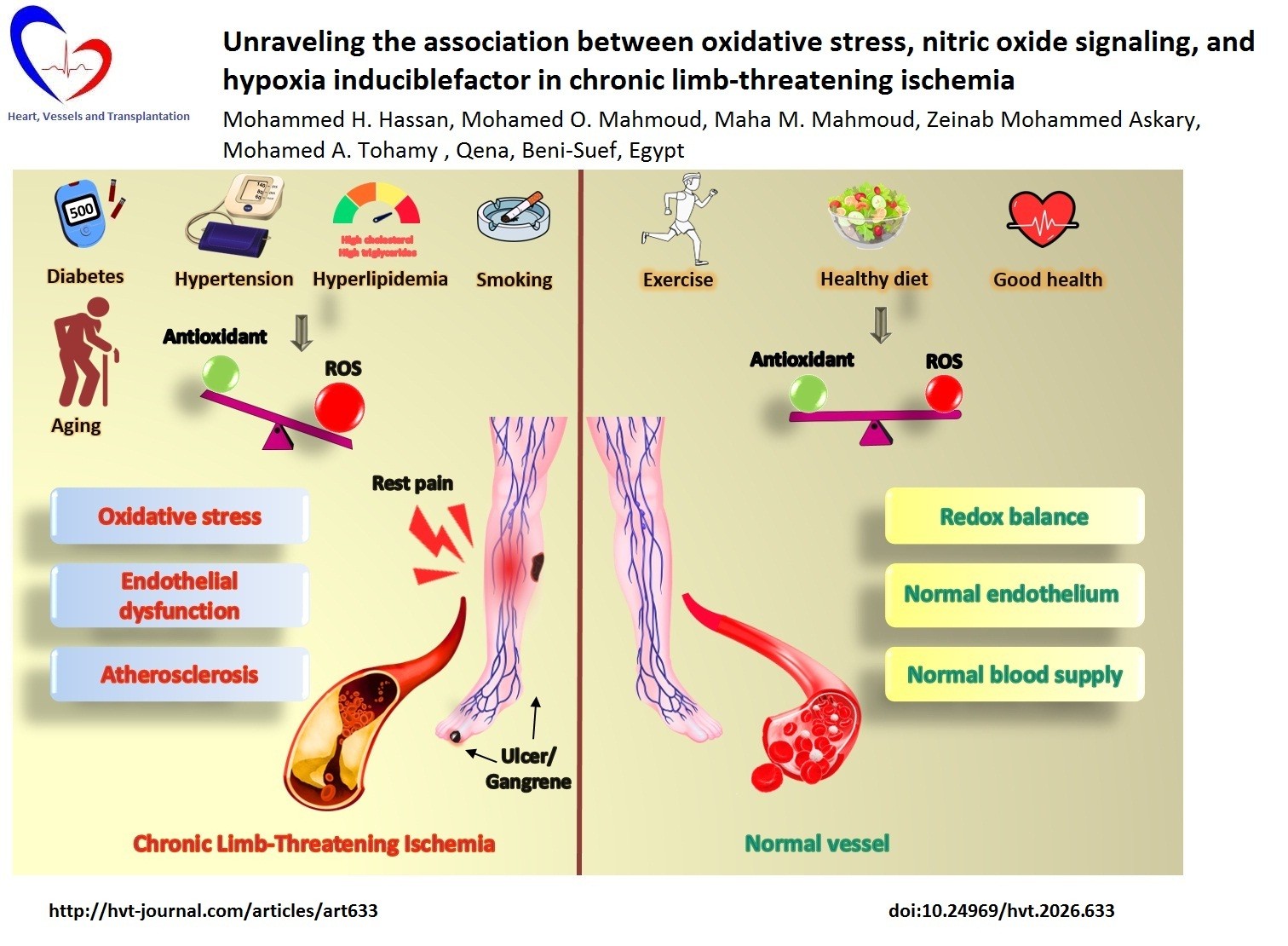
Graphical abstract: Significance of reactive oxygen species in vascular function (This figure was created by Canva.com)
Introduction
Chronic limb-threatening ischemia (CLTI), a severe clinical subgroup of peripheral arterial disease (PAD) is characterized by ischemic rest discomfort, non-healing ulcers, or gangrene (1). Using CLTI-specific ICD-10-CM coding, an updated study on the incidence and prevalence of CLTI in the Medicare population found that the estimated yearly incidence was 0.33%, while the estimated 2-year prevalence was 0.74% (2).
Patients who have undergone surgery for CLTI show that in comparison to males, women are underrepresented and have fewer risk factors and comorbidities. However, CLTI targets women above the age of 75 (3). In order to diagnose CLTI, ischemic rest discomfort or tissue loss must be present together with an established PAD. The presence of pain should last longer than two weeks and be linked to at least one abnormal hemodynamic parameter, such as flat or minimal pulsatile volume recording (PVR) waveforms, ankle brachial index < 0.4, absolute AP < 50 mmHg, absolute toe pressure < 30 mmHg, and transcutaneous pressure of oxygen < 30 mmHg (4). Patients with small vascular disease caused by diabetes mellitus, hypertension, or chronic renal disease have lower ankle-brachial pressure index sensitivity, and vascular stiffness is less likely to affect the toe vessels, so in this case, the toe-brachial index (TBI) can be employed, where TBI < 0.7 is considered an abnormal result (5). For CLTI classification, there are many classification guidelines; however, the Fontaine and Rutherford categories are most frequently used by vascular experts to classify the severity of CLTI, whereas the WIfI category evaluates the risk of lower extremity amputation based on the size of the wound, the degree of ischemia, and the existence of a foot infection (6). Although CLTI occurs in just 11 to 20% of all documented PAD cases, it is very serious, with a reported 15-30% 1-year limb amputation rate and 10-25% mortality rate (7). Therefore, further research into illness triggers and the development of novel treatment options is crucial.
Free radicals, including the reactive oxygen species (ROS), reactive nitrogen species (RNS), and sulfur reactive species, do not necessarily represent harmful substances; they play vital physiological roles as mediators in cellular communication and immune responses, on condition that the equilibrium between the amount of free radicals and the effectiveness of antioxidant defenses is maintained. Once this equilibrium is disrupted, a state of oxidative stress occurs, leading to the destruction of deoxyribonucleic acid (DNA), proteins, and lipids (8, 9). Endothelial dysfunction, the first stage in the pathophysiology of atherosclerosis, which can be driven by oxidative stress, is associated with predisposing risk factors such as diabetes mellitus, hypertension, hypercholesterolemia, and smoking, resulting in many cardiovascular issues (10). Oxidative stress can lead to endothelial dysfunction through a number of mechanisms, including inducing endothelial cell (EC) apoptosis, increasing EC adhesion to monocytes, altering EC angiogenesis potential, ROS-triggered inflammation, ROS-triggered mitochondrial dysfunction, and compromising nitric oxide (NO) endothelium-dependent vasorelaxation (11, 12). The latter process is the key pathway that we will focus on.
Endothelial nitric oxide synthase (eNOS), the most common NOS isoform in the vasculature and responsible for the majority of NO generated within tissue, causes dilation of all types of blood vessels by activating soluble guanylyl cyclase and raising cyclic guanosine monophosphate (cGMP). NO is an effective suppressor of platelet aggregation and adhesion, as well as leukocyte adhesion; therefore, it can defend against atherogenesis and fibrous plaque development in the vasculature (13, 14). An imbalance between NO production and ROS generation triggers impairment of vessel dilation, inflammatory responses, platelet aggregation, and smooth muscle cell proliferation, which impairs blood and oxygen delivery (15).
The majority of multicellular creatures have developed molecular processes that allow them to adapt to hypoxic circumstances, which occur when oxygen demand exceeds supply. These mechanisms involve the activation of genes that generate proteins that improve oxygen supply and modify the metabolic process in a hypoxic tissue (16). The hypoxia-inducible factor (HIF), a transcriptional regulatory protein, is one of the main cellular adaptive mechanisms (17). A family of HIF-dependent adaptive genes, including those involved in the regulation of angiogenesis, metabolic processes, erythropoiesis, and vascular tone, is coordinated by HIF accumulation driven by hypoxia. Increased blood and oxygen flow to the ischemic tissue is the outcome of these genes' expression (18).
This review evaluating the relationship between oxidative stress, NO, and hypoxia-HIF in CLTI aims to summarize how these interrelated pathways contribute to disease pathogenesis and prospective therapeutics.
Methods
The electronic databases PubMed, Google Scholar, E-library, and the websites of medical journals (medicine, biology, and biochemistry) represented the literature search. The following search terms were used: oxidative stress, chronic limb-threatening ischemia, chronic limb ischemia, critical limb ischemia, endothelial dysfunction, nitric oxide, hypoxia inducible factor.
Articles published between 2010 and 2025 met the requirements for inclusion in the narrative review. Articles about the connection between oxidative stress, NO, and hypoxia-HIF in CLTI were among the literary references. Articles without full text, significant content, or about acute lower limb ischemia were excluded from consideration. Overall, 214 items were found during the data search. After preliminary screening to ensure they aligned with the research's objectives, 109 were chosen for additional examination as a result.
Oxidative stress: the major cause of CLTI
In the majority of cells, mitochondria have been the main source of intracellular oxidant generation, followed by the nicotinamide adenine dinucleotide phosphate (NADP) oxidases (NOXs), xanthine oxidase, and heme oxygenase 1 sources. Overproduction or buildup of ROS, such as superoxide, hydroxyl, and peroxyl radicals, as well as hydrogen peroxide and hypochlorous acid, triggers a state of oxidative stress (19). ROS are crucial for the oxidation of low-density lipoprotein (LDL), inflammatory responses, and changes in vascular tone. They also raise the expression of cell adhesion molecules on endothelial cells, such as P and E-selectins and vascular cell adhesion molecules-1 (VCAM-1), which attract leukocytes into the inner endothelial space (20). The loosening of the endothelial membrane facilitates the invasion of monocytes and oxidized LDL (oxLDL) from the vessel lumen to its wall. After leaving the vessel's lumen, monocytes transform into macrophages and take up oxLDL, becoming foam cells. Over time, foam cells disintegrate and form the plaque's lipid core (21). The fibrous section, including the stabilizing cap, is formed by vascular smooth muscle cells (VSMCs) that are triggered by chemokines and adhesion factors. As these cells migrate to the site of inflammation, they generate collagen and elastin, which help to solidify the formed atherosclerotic plaque (22) (Fig. 1). When the endothelium wears down or a plaque layer cracks, a chain reaction develops, leading to platelet-rich thrombus development and further narrowing of the blood vessel lumen (23). Restricted flow of blood in peripheral tissues results in diminished oxygenation and supply to peripheral tissues, leading to claudication or rest discomfort, which develops into chronic limb ischemia (24).

Figure 1. Oxidative stress triggers the formation of atherosclerotic plaque (This figure was created by Canva.com)
ICAM-1 – intercellular cell adhesion molecule 1, oxLDL – oxidized low-density lipoprotein, ROS-reactive oxygen species, VCAM1- vascular cell adhesion molecule-1
Therefore, many variables, including endothelial dysfunction, leukocyte activation and inflammation, and altered microvascular flow due to oxidative stress, all affect the final clinical picture of CLTI (25).
Oxidative stress and NO bioavailability
NO generation is the primary mechanism by which the endothelium sustains its defense function against vascular disorders (26). As a biologically vasoactive substance, NO is involved in blood pressure control, vascular permeability regulation, vasodilation, and blood flow-dependent dilatation (27). Various cofactors are necessary for NOS to produce NO from its substrate L-arginine, including tetrahydrobiopterin (BH4). NOS uncoupling is the process by which the oxygenase domain of NOS monomers produces superoxide anions rather than NO in the lack of BH4 or its oxidation to BH3 or BH2 and L-arginine deficiency (28). Furthermore, excessive ROS levels may readily inactivate NO to produce peroxynitrite, which lowers the quantity of functional NO. Importantly, peroxynitrite is a potent oxidant that promotes atherogenesis and has the ability to uncouple eNOS, increasing superoxide and decreasing NO bioavailability (29) (Fig. 2). The excessive level of generated ROS can restrict vascular development and remodeling by generating endothelial dysfunction, which includes decreased vasomotor activity, monocyte infiltration, poor endothelial barrier integrity, and increased risk of thrombosis (30, 31). Increasing stenosis may compromise the adaptive ability of arteriogenesis, leading to degradation of newly formed arterial frameworks and exacerbating muscle cell destruction. Physical activity has been limited owing to ischemic discomfort, and further diminishing exercise capacity occurs (32).

Figure 2. The impact of oxidative stress on nitric oxide availability. A: The eNOS can synthesize NO from its precursor in the presence of its cofactor. B: In oxidative stress condition eNOS undergoes uncoupling and forms superoxide instead of NO. (This figure was created by Canva.com)
Arg – arginine, BH – tetrahydrobiopterin, CaM -calmodulin, eNOS - endothelial nitric oxide synthase, FAD – flavine adenine dinucleotide, FMN- flavine mononucleotide, NADP –oxidized nicotinamide adenine dinucleotide phosphate, NADPH –reduced nicotinamide adenine dinucleotide phosphate, NO – nitric oxide, ROS-reactive oxygen species
NO production can be impacted by NOS polymorphisms that alter NOS expression or activity. Although NO is generated by neuronal (NOS1), inducible (NOS2), and endothelial (NOS3) nitric oxide synthases, NOS3 is the main isoform for NO production in the circulatory system. The NOS3 gene has several variation regions, such as insertions or deletions of nucleotides, single-nucleotide polymorphisms (SNPs), and variable numbers of tandem repeats (VNTRs) (33). The 7q35–7q36 locus of human chromosome 7 contains the NOS3 gene (34). The SNP database contains over 1700 genetic variations, some of which are recognized as effective because they impact NOS3 transcription or function, such as the SNPs rs2070744, rs1799983, rs3918226, and a VNTR in intron 4, which have been extensively researched (35). The SNP rs1799983 is found in exon 7 and is referred to as Glu298Asp because a guanine is altered to thymine at position 894 of the NOS3 gene, which results in a substitution of glutamine (Glu) to aspartate (Asp) at position 298 of the protein (36). According to Joshi et al. (37), endothelial cells with the variation of Asp allele for the Glu298Asp polymorphism showed reduced NOS3 activity. The SNP rs2070744, often known as the -786T>C SNP, has cytosine for the substitution of thymine at location 786 of the NOS3 promoter (38). Miyamoto et al. (39) found that people with the –786T→C mutation had considerably lower serum nitrite/nitrate levels than people without the mutation (i.e., carriers of the T allele), indicating that this SNP decreases NOS3 transcriptional activity. The SNP rs3918226 (665C>T) involves the replacement of C with T in the promoter region (40). This alteration reduces promoter function by 20–40%, which was proved by Salvi et al. (41) using HeLa and HEK293T cells transfected with the NOS3 promoter incorporating either the C or the T allele at position 665 in the NOS3 gene. The eNOS gene's 4b/4a VNTR polymorphism in intron 4 (varying number of tandem repeats on intron 4) controls eNOS after transcription by modifying the production of a small interfering ribonucleic acid (siRNA). The most frequently identified alleles of this polymorphism are those containing four (variant 4a) or five (variant 4b) copies of the 27 bp siRNA sequence (42). According to research by Zhang et al. (43), endothelial cells with variant 4b exhibit higher amounts of sirRNA than cells with variant 4a, which results in a decrease of NOS3 mRNA.
Oxidative stress and HIF1 bioavailability
When oxygen levels in the cell microenvironment are lower than the usual physiological condition, numerous physiological and pathological processes happen (44). HIF is an essential regulatory protein that maintains oxygen homeostasis and is primarily responsible for maintaining the equilibrium between oxygen demand and supply (18). A constitutive β subunit plus one of the three oxygen-dependent α subunits, HIF-1α, HIF-2α, or HIF-3α, constitute HIFs (45). In normoxia, certain prolyl hydroxylases (PHDs) utilize O2 as a substrate to hydroxylate HIF-α. When HIF-α is hydroxylated, it can connect with von Hippel–Lindau protein and attract ubiquitin ligase, which causes HIF-α to be degraded by proteases. When PHD activity is suppressed in hypoxic environments, HIF-α builds up, diffuses to the nucleus, and combines with the HIF-β subunit to produce HIF-1, HIF-2, or HIF-3 (46). Factors inhibiting HIF are additional HIF-α controlling enzymes that facilitate an asparagine hydroxylation process that inhibits the binding of HIF-α with CBP/p300 transcriptional co-activating factors (47) (Fig.3).
After that, HIF promoting angiogenesis by upregulating growth factors like VEGF and enabling an energy-saving transition from aerobic to anaerobic metabolism by upregulating important glycolytic enzymes (48). Although HIF activation increases tissue oxygenation and angiogenesis, during chronic ischemia its adaptive function becomes inappropriate, worsening tissue damage by producing more ROS. The cytochrome chain, which is primarily controlling mitochondrial oxidative phosphorylation, may be altered by HIF-1, leading to decreased adenosine triphosphate synthesis and increased ROS production (49, 50). Furthermore, HIF-1 increases the production of the reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX) class, especially NOX1 and NOX4. These NOX subtypes increase oxidative stress by producing ROS (51).
Due to its primary function in many cells, the research specifically focused on HIF1α polymorphism and concluded that polymorphisms could impact the HIF1α mRNA or protein expression and stability (52, 53). Chromosome 14 (q21-24) encodes the human HIF-1α gene, which is subject to several single-nucleotide polymorphisms; thereby, the structure and biological activity of the HIF-1α gene can be affected (54). C1772T (rs11549465), G1790A (rs11549467), C111A, and rs2057482 in UTR are considered the most widely researched SNPs (55). For example, Kim et al. (56) reported that the C1772T polymorphism had been associated with elevated expression of HIF-1α. According to Fraga et al. (57), higher production of the HIF-1α protein is linked to the C1772T and G1790A SNPs of the HIF-1α gene.

Figure 3. Correlation between oxygen status and hypoxia inducible factor 1 (This figure was created by Canva.com)
FIH -factors inhibiting hypoxia inducible factor, HIF- hypoxia inducible factor, PHD- prolyl hydroxylases, pVHL- von Hippel–Lindau protein
Koukourakis et al. (58) investigated the impact of the C2028T polymorphism of the HIF-1α on the protein expression and found a positive correlation.
Additionally, several HIF-1α genetic polymorphisms, including rs10873142 (C>T) in intron 8, rs41508050 (C>T) in exon 10, rs2783778 (C>T) and rs7148720 (T>C) in 5’-flanking, rs11549465, (C>T), and rs11549467 (G>A) in exon 12, and rs2057482 (T>C) 3’in UTR region, have been linked to cardiovascular diseases such as ischemic heart disease, acute myocardial infarction, and coronary artery disease (59).
Oxidative stress risk factors
Numerous earlier studies have shown that conditions including diabetes mellitus, high blood pressure, and high cholesterol increase the production of ROS, which causes oxidative stress (60), which will be clarified in the following section (Fig. 4).
Diabetes mellitus
In hyperglycemia, nonenzymatic interactions between monosaccharides, such as glucose, glyceraldehyde, and fructose, and amino groups of proteins, lipids, and nucleic acids can produce advanced glycation end products (AGEs) (61). ROS are produced when these AGEs bind with their receptors, causing oxidative stress via triggering of protein kinase C (PKC), which increases NADPH oxidase and lipoxygenase (62). As a result of the inhibition of antioxidant systems caused by this oxidative stress, DNA damages induce repair enzymes like poly-ADP ribose polymerase-1, which inactivate glyceraldehyde-3-phosphate dehydrogenase. This inactivation leads to glyceraldehyde-3-phosphate, glucose 6-phosphate, and fructose 6-phosphate accumulation (63). This accumulation results in activation of alternative undesirable pathways, such as the polyol pathway, where the enzyme aldose reductase breaks down excess glucose into sorbitol and fructose, which worsens oxidative stress by reducing NADPH, which is an essential ingredient for the production of reduced glutathione (GSH) (64). All of these mechanisms lead to boosting oxidative stress state.

Figure 4. Oxidative stress risk factors associated with CLTI (This figure was created by Canva.com)
CLTI - chronic limb-threatening ischemia
Hyperlipidemia
Increased cholesterol results in modifications to the cell membrane's structure, which may make it easier for ROS to escape from the mitochondrial electron chain and activate NADPH oxidase. Lipid peroxidation in the cell membrane is caused by these reactive free radicals, which also produce additional free radicals (65). oxLDL suppresses mitochondrial activity and increases the production of mitochondrial ROS, which is implicated in LDL oxidation, leading to a continuous cycle (66). Excess LDLs cause oxidative stress by lowering antioxidant systems like superoxide dismutase, glutathione, and catalase , and elevating the activity of ROS-producing enzymes such as myeloperoxidase (67). High-density lipoprotein (HDL) cholesterol is known to have strong anti-inflammatory and antioxidant effects, as well as the potential to inhibit cholesterol transport and encourage cholesterol efflux from macrophages. HDL function depends on the phospholipid monolayer on its surface. OxLDLs are an effective provider of phospholipid oxidation, which leads to HDL inactivation (68). HDL levels are frequently lower in hyperlipidemia, and they also exhibit altered structure and decreased efficiency (69).
Hypertension
An elevation in blood pressure within the blood vessels induces a biological stress response, which is transferred to the intracellular region via different sensor systems, and it has been contributed to the elevated generation of ROS, which serves as the cause for vascular dysfunction (70). Angiotensin II (Ang II) significantly increases the function of the NADPH oxidase enzyme, which leads to the generation of superoxide anion via the AT1 receptor. Long-term Ang II activation also increases the production of certain NOX components, such as p22 phox and p47 phox, and reduces the expression of several scavenging proteins in the cell, which raises intracellular ROS levels. Also, elevated ROS levels may promote the expression of renin-angiotensin-aldosterone system factors that prefer Ang II, creating a potentially damaging feedback loop (71, 72). Furthermore, PKC and Rho family guanosine triphosphatases (GTP), such as Rac1, are phosphorylated and activated during Ang II stimulation. These processes are necessary for the formation of the functional NOX molecule at the cell membrane (73). Elevated aldosterone can also cause oxidative stress through a variety of mechanisms, such as activating NADPH oxidase and uncoupling of NOS (74). An overproduction of unfolded and misfolded proteins in the endoplasmic reticulum (ER) lumen, known as ER stress, is becoming recognized as a potential contributor to hypertension, which leads to the accumulation of ROS and oxidative stress. Additionally, mitochondrial malfunction and an increase in mitochondrial ROS production can be driven by ER stress (75). Additionally, decreases in glutathione peroxidase and superoxide dismutase activity have been reported in hypertensive people, increasing the oxidative stress state (76).
Aging
The oxidative stress theory of aging relies on the structural damage due to the generation of oxidative degradation of macromolecules such as DNA, lipids, and proteins via RNS/ROS, resulting in age-related biological deficits (77). Oxidative stress is a result of a variety of modifications caused by age, including mitochondrial malfunction and a decline in the effectiveness of different physiological antioxidant defense mechanisms (78). The existence of a smaller percentage of l-arginine in the cytoplasm of endothelial cells to be used as an eNOS substrate is one of the mechanisms causing the uncoupling of eNOS and diminished NO release with aging. This is because arginase, an enzyme that breaks down L-arginine, is more expressed and active in elderly individuals (79). Aging-related insufficient autophagy is thought to be another factor contributing to a rise in oxidative stress, which builds up damaged proteins and encourages more oxidative stress along with inflammation (80). Telomere shortening is a significant biomarker of aging that activates the DNA damage response. The continuous triggering of these damage pathways causes cells to enter senescence or apoptosis, as well as inducing a pro-inflammatory response, which exacerbates oxidative stress within the cell. It can often be associated with mitochondrial malfunction, leading to the overproduction of ROS (81).
Smoking
Numerous smoking-related illnesses are attributed to systemic oxidative stress, and smoking is recognized to be a cause and a promoter of oxidative stress (82). Cigarette smoke contains thousands of compounds that can induce cellular oxidative stress, including quinones, reactive aldehydes, polycyclic hydrocarbons, and reactive oxygen and nitrogen species (ROS/RNS) (83). It has been demonstrated that smoking tobacco cigarettes significantly raises NOX2 activity, which is responsible for the increased production of superoxide in the vasculature (84). Cigarette smoking partially depletes cellular BH4 by impeding its de novo production via degradation of its rate-limiting enzyme GTP cyclohydrolase (GTPCH) by the ubiquitin proteasomal system (UPS). This leads to eNOS uncoupling and subsequently the generation of peroxynitrite anion (83, 85). The tobacco smoke tar phase compounds reduced molecular oxygen to produce O-2, which can then be converted into other free radicals such as H2O2 and OH. In addition, metals in cigarettes, such as chromium, nickel, iron, and copper, generate ROS via Fenton-like reactions, causing oxidative stress, whereas metals such as arsenic, cadmium, and mercury have an indirect effect on the antioxidant response by hindering GSH function, lowering its availability for the cellular antioxidant system. This creates an additional load of ROS (86). In addition to GSH, important endogenous and exogenous antioxidants like vitamin C (ascorbic acid), carotene, glutathione peroxidase, and superoxide dismutase are all downregulated by cigarette smoke (87). Furthermore, in the vapor part, Acrolein can stimulate the development of free radicals in endothelial cells (82).
Therapeutic potential and clinical implications
While conventional management options, such as endovascular or open surgical revascularization and angioplasty, are established treatments for CLTI (88, 89), adjunctive therapies targeting underlying mechanisms are warranted.
Focusing on reducing oxidative damage, enhancing NO-driven angiogenesis, and leveraging HIF-1’s role in promoting tissue oxygenation and vascular growth may have a potential outcome for improvement of the CLTI clinical outcomes.
Numerous antioxidants have shown encouraging results in lowering oxidative stress and enhancing CLTI outcomes. For instance, Lejay et al. (90) showed that N-acetyl cysteine therapy, which is thought to be a free radical scavenger by preserving an abundance of the rate-limiting cysteine precursor for glutathione antioxidant, permitted functional enhancement and restored mitochondrial activity in CLTI. Kuroda et al. (91) indicated that the application of Edaravone decreased oxidative stress in the ischemia-reperfusion limb. Furthermore, prior research has shown that therapy for hind limb ischemia with the significant antioxidants ellagic acid, berberine, and coenzyme Q10 can reduce lipid peroxidation byproducts and boost antioxidant enzyme activity (92, 93). Furthermore, Karas et al. (94) demonstrated that selenium nanoparticles have a significant capacity for capturing free radicals, resulting in faster wound healing. They also increase endogenous antioxidant enzyme activity by raising GSH and the mRNA expression of their associated enzymes, superoxide dismutase, and catalase.
The use of exogenous NO and activating eNOS has proved to be useful in increasing angiogenesis and arteriogenesis under critical ischemia conditions (95). According to Lessiani et al. (96), individuals with CLTI who use Iloprost had considerably higher levels of plasma nitrate and nitrite as well as significantly lower levels of endothelial dysfunction and oxidative stress. Moreover, as reported by Morita et al. (97), supplementing with l-arginine and l-citrulline increased plasma l-arginine levels more quickly and significantly improved NO bioavailability and plasma cGMP level in atherosclerotic rabbits. Statins were indicated to enhance the expression of the NOS3 gene and eNOS activity by enhancing the levels of BH4 via activation of its rate-limiting enzyme GTP cyclohydrolase 1 or inhibiting its oxidation (98). Additionally, naftidrofuryl, a 5-hydroxytryptamine type 2inhibitor, and cilostazol, a phosphodiesterase III inhibitor, are regarded as NO donors, as both can raise NOS expression, which triggers NO production in PADs (99). Efficient NO-releasing systems have been used to transport NO donors to particular areas, bypassing the limitations of NO's short lifespan and its random delivery. For example, Wijaya et al. (100) developed PLGA nanomaterial loaded with L-arginine and lovastatin to accomplish long-term release and increase NO production, exhibiting effective outcomes for atherosclerosis treatment. Ma et al. (101) constructed a nanosystem with porous cerium oxide nanoparticles loaded with L-arginine. The released L-arginine has been transformed into NO, which has a high potential for improving wound healing (101). Furthermore, Hou et al. (102) developed a NO delivery system relying on an enzyme-prodrug pair, galactosidase-galactosyl-NO, which has significantly boosted its therapeutic efficiency in the management of ischemic disorders and eliminated adverse effects related to the systemic release of NO. Cell therapy can also be employed to enhance NO production; for example, Madaric et al. (103) employed stem cell therapy in patients with CLTI and showed that the implemented bone marrow-derived mononuclear cells were responsible for decreased levels of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, resulting in a rise of NO and a reduction of oxidative stress. Using tissue engineering techniques, Zhang et al. (104) constructed vascular grafts using eNOS-modified MSCs implanted and grown on electrospun tubular scaffolds to generate artificial vessels that generated high eNOS enzyme and NO and might be effective in blood vessel regeneration.
Because HIF-1α plays a fundamental role in stimulating angiogenesis during hypoxic circumstances, it has been thoroughly investigated as a potential therapy for CLTI (48). Botusan et al. (105) found that dimethyloxalylglycine, a competitive antagonist of α-ketoglutarate, a cofactor for α-KG-dependent dioxygenases, including prolyl hydroxylase, can inhibit prolyl hydroxylase function and enhance HIF-1α stabilization, implying tissue regeneration and wound healing. Furthermore, several recent studies suggest that gene therapy might be utilized to boost HIF-1 levels and alleviate ischemia effects. For example, Vincent et al. (106) found that injecting a rabbit model of hindlimb ischemia with the HIF-1α/VP16 hybrid gene resulted in improved blood flow to affected regions. In a study by Rajagopalan et al. (107), individuals with PAD and CLTI had pain reduction and ulcer healing after receiving a single inramuscular injection of an adenoviral vector of HIF-1α.
In a diabetic model of CLTI, Sarkar et al. (108) demonstrated that an adenovirus encoding a constitutively active form of HIF-1α improved the recovery of limb perfusion and performance, restored the function of angiogenic cells, enhanced eNOS activity, and increased vessel density in the ischemic limb. HIF-α activity can also be reestablished through cell-based therapy.
For instance, in a mouse hind limb ischemia model, Pei et al. (109) indicated that transfecting HIF-1α into scaffold-based cardiac stem cells (CSCs) and introducing them to ischemic tissue resulted in enhanced circulation with muscle regeneration, suggesting that HIF-CSCs may be a potential alternative treatment for ischemic tissue reconstruction.
Gaps in current knowledge
There are few studies that explicitly look at the oxidative stress-NO-HIF tripartite interaction in human CLTI tissues; the majority of the information comes from general PAD or animal models of hind limb ischemia. Therefore, there is a need for models that replicate the complicated, contemporaneous interactions of ROS, NO, and HIF in order to create successful multi-target treatments. In addition, gaps include revascularization testing with NO donors, HIF stabilizers, or oxidative stress scavengers. Furthermore, multivariate research is necessary since comorbidities like diabetes influence results.
Future perspectives
CLTI patients show a wide range of differences in oxidative stress levels, microvascular dysfunction, and hypoxia signaling, making their condition highly heterogeneous. Incorporating advanced omics technologies such as transcriptomics, metabolomics, and redox proteomics, and integrating cutting-edge experimental platforms such as organoids and organ-on-chip technologies can help create personalized disease profiles for each patient. Combining these molecular insights with biochemical markers, imaging results, and clinical data through predictive algorithms can enhance diagnostic precision and treatment planning. Such strategies hold the promise of transforming CLTI management into a more personalized, effective approach tailored to individual patient needs.
Conclusion
In summary, oxidative stress, nitric oxide insufficiency, and hypoxia-inducible factor signaling interact intricately to cause chronic limb-threatening ischemia. Increased oxidative stress reduces the bioavailability of nitric oxide, thereby worsening endothelial dysfunction. Despite stabilizing HIF in this hypoxic environment, which encourages compensatory mechanisms like angiogenesis, these adaptations are not enough to mitigate ischemic tissue damage. Comprehending and focusing on these oxidative pathways, reestablishing nitric oxide signaling, and adjusting HIF activity have considerable therapeutic promise for enhancing CLTI results in clinical trials.
Peer-review: External and internal
Conflict of interest: None to declare
Authorship: M.H.H. - conceptualization; supervision; writing - review & editing, M.O.M.-conceptualization; supervision; writing - review & editing, M.M.M. - Writing - original draft, Z.M.A. - supervision; writing - review & editing, M.A.T. - conceptualization; supervision; writing - review & editing. The revised final paper was read and approved by each author. Thus all authors equally contributed to preparation of manuscript and fulfilled all authorship criteria
Acknowledgements and funding: None to declare
Statement on A.I.-assisted technologies use: The authors used AI-supported technologies in the preparation of figures .
Data and material availability: The study described in the paper did not utilize any data
References
| 1. Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FGR. Inter-society consensus for the management of peripheral arterial disease (TASC II). J Vasc Surg 2007; 45 (1 Suppl): S5-S67. doi:10.1016/j.jvs.2006.12.037 https://doi.org/10.1016/j.jvs.2006.12.037 PMid:17223489 |
||||
| 2. Kwong M, Rajasekar G, Utter GH, Nuño M, Mel, MW. Updated estimates for the burden of chronic limb-threatening ischemia in the medicare population. J Vasc Surg 2023; 77: 1760-75. doi:10.1016/j.jvs.2023.01.200 https://doi.org/10.1016/j.jvs.2023.01.200 PMid:36758910 |
||||
| 3. Martelli E, Zamboni M, Sotgiu G, Saderi L, Federici M, Sangiorgi GM, et al. Sex-related differences and factors associated with peri-procedural and 1-year mortality in chronic limb-threatening ischemia patients from the climate italian registry. J Person Med 2023; 13: 316. doi:10.3390/jpm13020316 https://doi.org/10.3390/jpm13020316 PMid:36836550 PMCid:PMC9959358 |
||||
| 4. Berchiolli R, Bertagna G, Adami D, Canovaro F, Torri, L, Troisi N. Chronic limb-threatening ischemia and the need for revascularization. J Clin Med 2023; 12: 2682. doi:10.3390/jcm12072682 https://doi.org/10.3390/jcm12072682 PMid:37048765 PMCid:PMC10095037 |
||||
| 5. Høyer C, Sandermann J, Petersen LJ. The toe-brachial index in the diagnosis of peripheral arterial disease. J Vasc Surg 2013; 58: 231-8. doi:10.1016/j.jvs.2013.03.044 https://doi.org/10.1016/j.jvs.2013.03.044 PMid:23688630 |
||||
| 6. Gornik HL, Aronow HD, Goodney PP, Arya S, Brewster LP, Byrd L, et al. 2024 ACC/AHA/AACVPR/APMA/ABC/SCAI/SVM/SVN/SVS/SIR/VESS guideline for the management of lower extremity peripheral artery disease: A report of the American College of Cardiology/American Heart Association joint committee on clinical practice guidelines. Circulation 2024; 149: e1313-e410. doi:10.1161/CIR.0000000000001251 https://doi.org/10.1161/CIR.0000000000001251 PMid:38743805 PMCid:PMC12782132 |
||||
| 7. Bloch RA, Ellias SD, Caron E, Shean KE, Prushik SG, Stone DH, et al The impact of follow-up on mortality in chronic limb-threatening ischemia. Ann Vasc Surg 2025; 112: 74-81. doi:10.1016/j.avsg.2024.11.092 https://doi.org/10.1016/j.avsg.2024.11.092 PMid:39675698 |
||||
| 8.Garcia-Llorens G, El Ouardi M, Valls-Belles, V. Oxidative stress fundamentals: Unraveling the pathophysiological role of redox imbalance in non-communicable diseases. Appl Sci 2025; 15: 10191. doi:10.3390/app151810191 https://doi.org/10.3390/app151810191 |
||||
| 9. Di Meo S, Venditti P. Evolution of the knowledge of free radicals and other oxidants. Oxid Med Cell Long 2020; 2020: 9829176. doi:10.1155/2020/9829176 https://doi.org/10.1155/2020/9829176 PMid:32411336 PMCid:PMC7201853 |
||||
| 10. Landmesser U, Hornig B, Drexler H. Endothelial function. Circulation 2004; 109(21 suppl 1): II-27-II-33. doi:10.1161/01.CIR.0000129501.88485.1f https://doi.org/10.1161/01.CIR.0000129501.88485.1f PMid:15173060 |
||||
| 11. Shaito A, Aramouni K, Assaf R, Parenti A, Orekhov A, Yazbi AE, et al. Oxidative stress-induced endothelial dysfunction in cardiovascular diseases. FBL 2022; 27: 105. doi:10.31083/j.fbl2703105 https://doi.org/10.31083/j.fbl2703105 PMid:35345337 |
||||
| 12. Taniyama Y, Griendling KK. Reactive oxygen species in the vasculature. Hypertension 2003; 42: 1075-81. doi:10.1161/01.HYP.0000100443.09293.4F https://doi.org/10.1161/01.HYP.0000100443.09293.4F PMid:14581295 |
||||
| 13. Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease. Circulation 2006; 113: 1708-14. doi:10.1161/CIRCULATIONAHA.105.602532 https://doi.org/10.1161/CIRCULATIONAHA.105.602532 PMid:16585403 |
||||
| 14. Förstermann U, Closs EI, Pollock JS, Nakane M, Schwarz P, Gath I, et al. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994; 23(6 pt 2): 1121-31. doi:10.1161/01.HYP.23.6.1121 https://doi.org/10.1161/01.HYP.23.6.1121 PMid:7515853 |
||||
| 15. Higashi Y, Maruhashi T, Noma K, Kihara Y. Oxidative stress and endothelial dysfunction: Clinical evidence and therapeutic implications. Trends Cardiovasc Med 2014; 24: 165-9. doi:10.1016/j.tcm.2013.12.001 https://doi.org/10.1016/j.tcm.2013.12.001 PMid:24373981 |
||||
| 16. Taylor CT, McElwain JC. Ancient atmospheres and the evolution of oxygen sensing via the hypoxia-inducible factor in metazoans. Physiol 2010; 25: 272-9. doi:10.1152/physiol.00029.2010 https://doi.org/10.1152/physiol.00029.2010 PMid:20940432 |
||||
| 17. Semenza GL , Agani F , Booth G , Forsythe J , Iyer N , Jiang B-H , et al. Structural and functional analysis of hypoxia-inducible factor 1. Kidney Internat 1997; 51: 553-5. doi:10.1038/ki.1997.77 https://doi.org/10.1038/ki.1997.77 PMid:9027737 |
||||
| 18. Lee P, Chandel, NS, Simon, MC. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nature reviews Mol Cell Biol 2020; 21: 268-83. doi:10.1038/s41580-020-0227-y https://doi.org/10.1038/s41580-020-0227-y PMid:32144406 PMCid:PMC7222024 |
||||
| 19. Valaitienė J, Laučytė-Cibulskienė A. Oxidative stress and its biomarkers in cardiovascular diseases. Artery Res 2024; 30: 18. doi:10.1007/s44200-024-00062-8 https://doi.org/10.1007/s44200-024-00062-8 |
||||
| 20. Kattoor AJ, Pothineni NVK, Palagiri D, Mehta JL. Oxidative stress in atherosclerosis. Curr Atherosc Rep 2017; 19: 42. doi:10.1007/s11883-017-0678-6 https://doi.org/10.1007/s11883-017-0678-6 PMid:28921056 PMCid:PMC12123089 |
||||
| 21. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circulation Res 2014; 114: 1852-66. doi:10.1161/CIRCRESAHA.114.302721 https://doi.org/10.1161/CIRCRESAHA.114.302721 PMid:24902970 PMCid:PMC8319769 |
||||
| 22. Alonso-Herranz L, Albarrán-Juárez J, Bentzon JF. Mechanisms of fibrous cap formation in atherosclerosis. Front Cardiovasc Med 2023; 10: 1254114. doi:10.3389/fcvm.2023.1254114 https://doi.org/10.3389/fcvm.2023.1254114 PMid:37671141 PMCid:PMC10475556 |
||||
| 23. Simon F, Oberhuber A, Floros N, Düppers P, Schelzig H, Duran M. Pathophysiology of chronic limb ischemia. Gefässchirurgie 2018; 23: 13-8. doi:10.1007/s00772-018-0380-1 https://doi.org/10.1007/s00772-018-0380-1 PMid:29950791 PMCid:PMC5997105 |
||||
| 24. Uccioli L, Meloni M, Izzo V, Giurato L, Merolla S, Gandini R. Critical limb ischemia: Current challenges and future prospects. Vasc Health Risk Manag 2018; 14: 63-74. doi:10.2147/VHRM.S125065 https://doi.org/10.2147/VHRM.S125065 PMid:29731636 PMCid:PMC5927064 |
||||
| 25. McArdle MJ, Giri J, Mohler III ER. Progression of peripheral artery disease to critical limb ischemia, in Critical limb ischemia: Acute and chronic. Springer. 2016: pp. 121-9. doi:10.1007/978-3-319-31991-9_13 https://doi.org/10.1007/978-3-319-31991-9_13 |
||||
| 26. Drummond GR, Sobey CG. Endothelial nadph oxidases: Which nox to target in vascular disease? Trends Endocrin Metab 2014; 25: 452-63. doi:10.1016/j.tem.2014.06.012 https://doi.org/10.1016/j.tem.2014.06.012 PMid:25066192 |
||||
| 27. Sessa W. Molecular control of blood flow and angiogenesis: Role of nitric oxide. J Thromb Haemost 2009. 7: 35-37. doi:10.1111/j.1538-7836.2009.03424.x https://doi.org/10.1111/j.1538-7836.2009.03424.x PMid:19630764 |
||||
| 28. Vanhoutte PM, Zhao Y, Xu A, Leung SW, Thirty years of saying no: Sources, fate, actions, and misfortunes of the endothelium-derived vasodilator mediator. Circulation Res 2016; 119: 375-96. doi:10.1161/CIRCRESAHA.116.306531 https://doi.org/10.1161/CIRCRESAHA.116.306531 PMid:27390338 |
||||
| 29. Förstermann U. Nitric oxide and oxidative stress in vascular disease. Pflügers Arch Eur J Physiol 2010; 459: 923-39. doi:10.1007/s00424-010-0808-2 https://doi.org/10.1007/s00424-010-0808-2 PMid:20306272 |
||||
| 30. Kumar P, Shen Q, Pivetti CD, Lee ES, Wu MH, Yuan SY. Molecular mechanisms of endothelial hyperpermeability: Implications in inflammation. Expert Rev Mol Med 2009; 11: e19. doi:10.1017/S1462399409001112 https://doi.org/10.1017/S1462399409001112 PMid:19563700 PMCid:PMC2828491 |
||||
| 31. Majno G. Chronic inflammation: Links with angiogenesis and wound healing. Am J Pathol 1998; 153: 1035. doi:10.1016/S0002-9440(10)65648-9 https://doi.org/10.1016/S0002-9440(10)65648-9 PMid:9777935 PMCid:PMC1853039 |
||||
| 32. Coats P, Jarajapu Y, Hillier C, McGrath J, Daly C. The use of fluorescent nuclear dyes and laser scanning confocal microscopy to study the cellular aspects of arterial remodelling in human subjects with critical limb ischaemia. Exp Physiol 2003; 88: 547-54. doi:10.1113/eph8802552 https://doi.org/10.1113/eph8802552 PMid:12861343 |
||||
| 33. Oliveira-Paula GH, Lacchini R, Tanus-Santos JE. Endothelial nitric oxide synthase: From biochemistry and gene structure to clinical implications of nos3 polymorphisms. Gene 2016. 575: 584-99. doi:10.1016/j.gene.2015.09.061 https://doi.org/10.1016/j.gene.2015.09.061 PMid:26428312 PMCid:PMC6728140 |
||||
| 34. Marsden PA, Heng H, Scherer S, Stewart R, Hall A, Shi X, et al. Structure and chromosomal localization of the human constitutive endothelial nitric oxide synthase gene. J Biol Chem 1993; 268: 17478-88. doi:10.1016/S0021-9258(19)85359-0 https://doi.org/10.1016/S0021-9258(19)85359-0 PMid:7688726 |
||||
| 35. Oliveira-Paula GH, Lacchini R, Tanus-Santos JE. Clinical and pharmacogenetic impact of endothelial nitric oxide synthase polymorphisms on cardiovascular diseases. Nitric Oxide 2017; 63: 39-51. doi:10.1016/j.niox.2016.08.004 https://doi.org/10.1016/j.niox.2016.08.004 PMid:27569446 |
||||
| 36. Luo Z, Jia A, Lu Z, Muhammad I, Adenrele A, Song Y. Associations of the NOS3 rs1799983 polymorphism with circulating nitric oxide and lipid levels: A systematic review and meta-analysis. Postgrad Med J 2019; 95: 361-71. doi:10.1136/postgradmedj-2019-136396 https://doi.org/10.1136/postgradmedj-2019-136396 PMid:31138610 |
||||
| 37. Joshi MS, Mineo C, Shaul, PW, Bauer JA. Biochemical consequences of the NOS3 glu298asp variation in human endothelium: Altered caveolar localization and impaired response to shear. FASEB 2007; 21: 2655. doi:10.1096/fj.06-7088com https://doi.org/10.1096/fj.06-7088com PMid:17449720 PMCid:PMC7460804 |
||||
| 38. Casas JP, Cavalleri GL, Bautista LE, Smeeth L, Humphries SE, Hingorani AD.Endothelial nitric oxide synthase gene polymorphisms and cardiovascular disease: A huge review. Am J Epidemiol 2006; 164: 921-35. doi:10.1093/aje/kwj302 https://doi.org/10.1093/aje/kwj302 PMid:17018701 |
||||
| 39. Miyamoto Y, Saito Y, Nakayama M, Shimasaki Y, Yoshimura T, Yoshimura M, et al. Replication protein a1 reduces transcription of the endothelial nitric oxide synthase gene containing a -786t→c mutation associated with coronary spastic angina. Hum Mol Genet 2000; 9: 2629-37. doi:10.1093/hmg/9.18.2629 https://doi.org/10.1093/hmg/9.18.2629 PMid:11063722 |
||||
| 40. Olivi L, Gu Y, Salvi E, Liu Y, Thijs L, Velayutham D, et al. The− 665 c> t polymorphism in the enos gene predicts cardiovascular mortality and morbidity in white europeans. J Human Hypert 2015; 29: 167-72. doi:10.1038/jhh.2014.66 https://doi.org/10.1038/jhh.2014.66 PMid:25102225 |
||||
| 41. Salvi E, Kuznetsova T, Thijs L, Lupoli S, Stolarz-Skrzypek K, D'Avila F, et al. Target sequencing, cell experiments, and a population study establish endothelial nitric oxide synthase (ENOS) gene as hypertension susceptibility gene. Hypertension 2013; 62: 844-52. doi:10.1161/HYPERTENSIONAHA.113.01428 https://doi.org/10.1161/HYPERTENSIONAHA.113.01428 PMid:24019403 |
||||
| 42. Cozma A, Fodor A, Orasan OH, Vulturar R, Samplelean D, Negrean V, et al. Pharmacogenetic implications of enos polymorphisms (glu298asp, t786c, 4b/4a) in cardiovascular drug therapy. In Vivo 2019; 33: 1051-8. doi:10.21873/invivo.11573 https://doi.org/10.21873/invivo.11573 PMid:31280192 PMCid:PMC6689342 |
||||
| 43. Zhang M-X, Zhang C, Shen YH, Wang J, Li X-N, Chen L, et al. Effect of 27nt small RNA on endothelial nitric-oxide synthase expression. Mol Biol Cell 2008; 19: 3997-4005. doi:10.1091/mbc.e07-11-1186 https://doi.org/10.1091/mbc.e07-11-1186 PMid:18614799 PMCid:PMC2526692 |
||||
| 44. Semenza GL. Life with oxygen. Science 2007; 318: 62-4. doi:10.1126/science.1147949 https://doi.org/10.1126/science.1147949 PMid:17916722 |
||||
| 45. Koh MY, Powis G. Passing the baton: The HIF switch. Trend Biochem Sci 2012; 37: 364-72. doi:10.1016/j.tibs.2012.06.004 https://doi.org/10.1016/j.tibs.2012.06.004 PMid:22818162 PMCid:PMC3433036 |
||||
| 46. Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010; 40: 294-309. doi:10.1016/j.molcel.2010.09.022 https://doi.org/10.1016/j.molcel.2010.09.022 PMid:20965423 PMCid:PMC3143508 |
||||
| 47. Zhang N, Fu Z, Linke S, Chicher J, Gorman JJ, Visk D, et al. The asparaginyl hydroxylase factor inhibiting HIF-1a3b1; is an essential regulator of metabolism. Cell Metabol 2010; 11: 364-78. doi:10.1016/j.cmet.2010.03.001 https://doi.org/10.1016/j.cmet.2010.03.001 PMid:20399150 PMCid:PMC2893150 |
||||
| 48. Carmichael E, Reme A-IS, Bosco PJ, Ortiz YY, Ramos DA, Gomez K, et al. Biological regulation of HIF-1α and its role in therapeutic angiogenesis for treatment of ischemic cardiovascular disease. Int J Mol Sci 2025; 26: 11236. doi:10.3390/ijms262211236 https://doi.org/10.3390/ijms262211236 PMid:41303720 PMCid:PMC12653304 |
||||
| 49. Millar NL, Reilly JH, Kerr SC, Campbell AL, Little KJ, Leach WJ, et al. Hypoxia: A critical regulator of early human tendinopathy. Ann Rheum Dis 2012; 71: 302-10. doi:10.1136/ard.2011.154229 https://doi.org/10.1136/ard.2011.154229 PMid:21972243 |
||||
| 50. Jassim AH, Fan Y, Pappenhagen N, Nsiah NY, Inman DM. Oxidative stress and hypoxia modify mitochondrial homeostasis during glaucoma. Antioxid Redox Sign 2021; 35: 1341-57. doi:10.1089/ars.2020.8180 https://doi.org/10.1089/ars.2020.8180 PMid:33736457 PMCid:PMC8817702 |
||||
| 51. Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Rad Biol Med 2016; 98: 18-28. doi:10.1016/j.freeradbiomed.2016.05.011 https://doi.org/10.1016/j.freeradbiomed.2016.05.011 PMid:27184955 PMCid:PMC4975970 |
||||
| 52. Tanimoto K, Yoshiga K, Eguchi H, Kaneyasu M, Ukon K, Kumazaki T, et al. Hypoxia-inducible factor-1α polymorphisms associated with enhanced transactivation capacity, implying clinical significance. Carcinogenesis 2003; 24: 1779-83. doi:10.1093/carcin/bgg132 https://doi.org/10.1093/carcin/bgg132 PMid:12919954 |
||||
| 53. Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Intern J Cancer 2016; 138: 1058-66. doi:10.1002/ijc.29519 https://doi.org/10.1002/ijc.29519 PMid:25784597 PMCid:PMC4573780 |
||||
| 54. Smolarz B, Łukasiewicz H, Samulak D, Kołaciński R, Langner S, Makowska M, et al. Hypoxia-induced factor-1α and its role in endometrial cancer. Anticanc Res 2024; 44: 3697-712. doi:10.21873/anticanres.17195 https://doi.org/10.21873/anticanres.17195 PMid:39197887 |
||||
| 55. Hu X, Fang Y, Zheng J, He Y, Zan X, Lin S, et al. The association between hif-1α polymorphism and cancer risk: A systematic review and meta-analysis. Tumor Biol 2014. 35: 903-16. doi:10.1007/s13277-013-1160-x https://doi.org/10.1007/s13277-013-1160-x PMid:24046090 |
||||
| 56. Kim HO, Jo YH, Lee J, Lee SS, Yoon KS. The c1772t genetic polymorphism in human hif-1α gene associates with expression of HIF-1α protein in breast cancer. Oncol Rep 2008; 20: 1181-7. doi:10.3892/or_00000127 https://doi.org/10.3892/or_00000127 |
||||
| 57. Fraga CADC, de Oliveira MVM, de Oliveira ÉS, Barros LO, Santos FBG, Gomez RS, et al. A high HIF-1α expression genotype is associated with poor prognosis of upper aerodigestive tract carcinoma patients. Oral Oncol 2012; 48: 130-5. doi:10.1016/j.oraloncology.2011.08.023 https://doi.org/10.1016/j.oraloncology.2011.08.023 PMid:21945343 |
||||
| 58. Koukourakis MI, Papazoglou D, Giatromanolaki A, Panagopoulos I, Maltezos E, Harris AL, et al. C2028t polymorphism in exon 12 and dinucleotide repeat polymorphism in intron 13 of the hif-1α gene define hif-1α protein expression in non-small cell lung cancer. Lung Cancer 2006; 53: 257-62. doi:10.1016/j.lungcan.2006.05.025 https://doi.org/10.1016/j.lungcan.2006.05.025 PMid:16837101 |
||||
| 59. Gladek I, Ferdin J, Horvat S, Calin GA, Kunej T. HIF1a gene polymorphisms and human diseases: Graphical review of 97 association studies. Genes Chrom Cancer 2017; 56: 439-52. doi:10.1002/gcc.22449 https://doi.org/10.1002/gcc.22449 PMid:28165644 PMCid:PMC5395341 |
||||
| 60. Sharifi-Rad M, Anil Kumar NV, Zucca P, Varoni EM, Dini L, Panzarini E, et al. Lifestyle, oxidative stress, and antioxidants: Back and forth in the pathophysiology of chronic diseases. Front Physiol 2020; 11: 552535. doi:10.3389/fphys.2020.00694 https://doi.org/10.3389/fphys.2020.00694 PMid:32714204 PMCid:PMC7347016 |
||||
| 61. An Y, Xu B-U, Wan S-R, Ma X-M, Long Y, Xu Y, et al. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc Diabetol 2023; 22: 237. doi:10.1186/s12933-023-01965-7 https://doi.org/10.1186/s12933-023-01965-7 PMid:37660030 PMCid:PMC10475205 |
||||
| 62. Twarda-Clapa A, Olczak A, Białkowska AM, Koziołkiewicz M. Advanced glycation end-products (ages): Formation, chemistry, classification, receptors, and diseases related to ages. Cells 2022; 11: 1312. doi:10.3390/cells11081312 https://doi.org/10.3390/cells11081312 PMid:35455991 PMCid:PMC9029922 |
||||
| 63. Rodríguez-Bolaños M, Perez-Montfort R. The remarkable role of triosephosphate isomerase in diabetes pathophysiology. Intern J Mol Sci 2025; 26: 8809. doi:10.3390/ijms26188809 https://doi.org/10.3390/ijms26188809 PMid:41009376 PMCid:PMC12470035 |
||||
| 64. Niimi N, Yako H, Takaku S, Chung SK, Sango K. Aldose reductase and the polyol pathway in schwann cells: Old and new problems. Intern J Mol Sci 2021; 22: 1031. doi:10.3390/ijms22031031 https://doi.org/10.3390/ijms22031031 PMid:33494154 PMCid:PMC7864348 |
||||
| 65. Singh, UN, Kumar S, Dhakal S. Study of oxidative stress in hypercholesterolemia. Int J Contemp Med Res 2017; 4: 1204-7. | ||||
| 66. Ballinger SW, Patterson C, Yan C-N, Doan R, Burow DL, Young CG, et al. Hydrogen peroxide-and peroxynitrite-induced mitochondrial DNA damage and dysfunction in vascular endothelial and smooth muscle cells. Circulation Res 2000; 86: 960-6. doi:10.1161/01.RES.86.9.960 https://doi.org/10.1161/01.RES.86.9.960 PMid:10807868 |
||||
| 67. Ganjali S, Keshavarz R, Hosseini S, Mansouri A, Mannarino MR, Pirro M, et al. Evaluation of oxidative stress status in familial hypercholesterolemia. J Clin Med 2021; 10: 5867. doi:10.3390/jcm10245867 https://doi.org/10.3390/jcm10245867 PMid:34945165 PMCid:PMC8707741 |
||||
| 68. Zerrad-Saadi A, Therond P, Chantepie S, Couturier M, Rye K-A, Chapman MJ, et al. Hdl3-mediated inactivation of ldl-associated phospholipid hydroperoxides is determined by the redox status of apolipoprotein ai and hdl particle surface lipid rigidity: Relevance to inflammation and atherogenesis. Arterioscler Thromb Vasc Biol 2009; 29: 2169-75. doi:10.1161/ATVBAHA.109.194555 https://doi.org/10.1161/ATVBAHA.109.194555 PMid:19762782 |
||||
| 69. Mollazadeh H, Carbone F, Montecucco F, Pirro M, Sahebkar A. Oxidative burden in familial hypercholesterolemia. J Cell Physiol 2018; 233: 5716-25. doi:10.1002/jcp.26466 https://doi.org/10.1002/jcp.26466 PMid:29323716 |
||||
| 70. Vecchione C, Carnevale D, Di Pardo A, Gentile MT, Damato A, Cocozza G, et al. Pressure-induced vascular oxidative stress is mediated through activation of integrin-linked kinase 1/βPIX/RAC-1 pathway. Hypertension 2009; 54: 1028-34. doi:10.1161/HYPERTENSIONAHA.109.136572 https://doi.org/10.1161/HYPERTENSIONAHA.109.136572 PMid:19770407 |
||||
| 71. Griendling KK, Minieri CA, Ollerenshaw JD, Alexander RW. Angiotensin ii stimulates nadh and nadph oxidase activity in cultured vascular smooth muscle cells. Circulation Res 1994; 74: 1141-8. doi:10.1161/01.RES.74.6.1141 https://doi.org/10.1161/01.RES.74.6.1141 PMid:8187280 |
||||
| 72. Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin ii within the cell nucleus. Hypertension 2010; 55: 166-71. doi:10.1161/HYPERTENSIONAHA.109.141622 https://doi.org/10.1161/HYPERTENSIONAHA.109.141622 PMid:19948986 PMCid:PMC2821807 |
||||
| 73. Tajima H, Naganishi S, Mukohda M, Ishida M, Hamada N, Shigemi N, et al. Critical role of the β isoform of protein kinase c (pKCβ) in angiotensin ii-induced oxidative stress in vascular smooth muscle cells. Physiol Rep 2025; 13: e70595. doi:10.14814/phy2.70595 https://doi.org/10.14814/phy2.70595 PMid:41103077 PMCid:PMC12531348 |
||||
| 74. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nature Rev Nephrol 2013; 9: 459-69. doi:10.1038/nrneph.2013.110 https://doi.org/10.1038/nrneph.2013.110 PMid:23774812 PMCid:PMC3922409 |
||||
| 75. Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Sign 2014; 21: 396-413. doi:10.1089/ars.2014.5851 https://doi.org/10.1089/ars.2014.5851 PMid:24702237 PMCid:PMC4076992 |
||||
| 76. Pedro-Botet J, Covas M, Martin S, Rubies-Prat J. Decreased endogenous antioxidant enzymatic status in essential hypertension. J Hum Hypertens 2000; 14: 343-5. doi:10.1038/sj.jhh.1001034 https://doi.org/10.1038/sj.jhh.1001034 PMid:10878691 |
||||
| 77. Liguori I, Russo G, Curcio F, Bulli, G, Aran L, Della-Morte D, et al. Oxidative stress, aging, and diseases. Clinical interventions in aging. 2018; 13: 757-72. doi:10.2147/CIA.S158513 https://doi.org/10.2147/CIA.S158513 PMid:29731617 PMCid:PMC5927356 |
||||
| 78. Buford TW. Hypertension and aging. Age Res Rev 2016; 26: 96-111. doi:10.1016/j.arr.2016.01.007 https://doi.org/10.1016/j.arr.2016.01.007 PMid:26835847 PMCid:PMC4768730 |
||||
| 79. Herrera MD, Mingorance C, Rodríguez-Rodríguez R, de Sotomayor MA. Endothelial dysfunction and aging: An update. Age Res Rev 2010; 9: 142-52. doi:10.1016/j.arr.2009.07.002 https://doi.org/10.1016/j.arr.2009.07.002 PMid:19619671 |
||||
| 80. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circulation Res 2018; 123: 825-48. doi:10.1161/CIRCRESAHA.118.312563 https://doi.org/10.1161/CIRCRESAHA.118.312563 PMid:30355078 PMCid:PMC6207260 |
||||
| 81. Huang X, Huang L, Lu J, Cheng L, Wu D, Li L, et al. The relationship between telomere length and aging-related diseases. Clin Exp Med 2025; 25: 72. doi:10.1007/s10238-025-01608-z https://doi.org/10.1007/s10238-025-01608-z PMid:40044947 PMCid:PMC11882723 |
||||
| 82. Fearon IM. Risk factors, oxidative stress, and cardiovascular disease. In: Laher I. (ed). Systems Biolology of Free Radicals and Antioxidants. Springer Berlin Heidelberg: Berlin, Heidelberg; 2014. pp. 765-90. doi:10.1007/978-3-642-30018-9_46 https://doi.org/10.1007/978-3-642-30018-9_46 |
||||
| 83. Abdelghany TM, Ismail RS, Mansoor FA, Zweier JR, Lowe F, Zweier JL. Cigarette smoke constituents cause endothelial nitric oxide synthase dysfunction and uncoupling due to depletion of tetrahydrobiopterin with degradation of gtp cyclohydrolase. Nitric Oxide 2018; 76: 113-21. doi:10.1016/j.niox.2018.02.009 https://doi.org/10.1016/j.niox.2018.02.009 PMid:29524646 |
||||
| 84. Carnevale R, Sciarretta S, Violi F, Nocella C, Loffredo L, Perri L, et al. Acute impact of tobacco vs electronic cigarette smoking on oxidative stress and vascular function. CHEST 2016; 150: 606-12. doi:10.1016/j.chest.2016.04.012 https://doi.org/10.1016/j.chest.2016.04.012 PMid:27108682 PMCid:PMC5408160 |
||||
| 85. Peluffo G, Calcerrada P, Piacenza L, Pizzano N, Radi, R. Superoxide-mediated inactivation of nitric oxide and peroxynitrite formation by tobacco smoke in vascular endothelium: Studies in cultured cells and smokers. Am J Physiol-Heart Circ Physiol 2009; 296: H1781-92. doi:10.1152/ajpheart.00930.2008 https://doi.org/10.1152/ajpheart.00930.2008 PMid:19363134 PMCid:PMC2716106 |
||||
| 86. Caliri AW, Tommasi S, Besaratinia A. Relationships among smoking, oxidative stress, inflammation, macromolecular damage, and cancer. Mutat Res 2021; 787: 108365. doi:10.1016/j.mrrev.2021.108365 https://doi.org/10.1016/j.mrrev.2021.108365 PMid:34083039 PMCid:PMC8287787 |
||||
| 87. Burke A, FitzGerald GA. Oxidative stress and smoking-induced vascular injury. Progr Cardiovasc Dis 2003; 46: 79-90. doi:10.1016/S0033-0620(03)00076-8 https://doi.org/10.1016/S0033-0620(03)00076-8 PMid:12920701 |
||||
| 88. Kinlay S. Management of critical limb ischemia. Circulation: Cardiovasc Interv 2016; 9: e001946. doi:10.1161/CIRCINTERVENTIONS.115.001946 https://doi.org/10.1161/CIRCINTERVENTIONS.115.001946 PMid:26858079 PMCid:PMC4827334 |
||||
| 89. Oddi FM, Granata G, Oddi L, Fedeli G, Brizzi V. Bridging the gaps: Insights from the 2024 esc guidelines on peripheral arterial and aortic diseases. Heart Vessels Transp 2024; 8: doi:10.24969/hvt.2024.530 https://doi.org/10.24969/hvt.2024.530 |
||||
| 90. Lejay A, Paradis S, Lambert A, Charles A-L, Talha S, Enache I, et al. N-acetyl cysteine restores limb function, improves mitochondrial respiration, and reduces oxidative stress in a murine model of critical limb ischaemia. Eur J Vasc Endovasc Surg 2018; 56: 730-8. doi:10.1016/j.ejvs.2018.07.025 https://doi.org/10.1016/j.ejvs.2018.07.025 PMid:30172667 |
||||
| 91. Kuroda Y, Togashi H, Uchida T, Haga K, Yamashita A, Sadahiro M. Oxidative stress evaluation of skeletal muscle in ischemia-reperfusion injury using enhanced magnetic resonance imaging. Scie Rep 2020; 10: 10863. doi:10.1038/s41598-020-67336-4 https://doi.org/10.1038/s41598-020-67336-4 PMid:32616815 PMCid:PMC7331576 |
||||
| 92. Tekin E, Kaya AK, Küçük A, Arslan M, Özer A, Demirtaş H, et al. Effects of ellagic acid and berberine on hind limb ischemia reperfusion injury: Pathways of apoptosis, pyroptosis, and oxidative stress. Medicina 2025; 61: 451. doi:10.3390/medicina61030451 https://doi.org/10.3390/medicina61030451 PMid:40142262 PMCid:PMC11943544 |
||||
| 93. Betul AY, Adoum BA. The investigation of the preventive effects of coenzyme q10 and berberine for tourniquet induced ischemia-reperfusion injury on skeletal muscle in rat hindlimb. GSC Biol Pharm Sci 2019. 9: 127-33. doi:10.30574/gscbps.2019.9.3.0238 https://doi.org/10.30574/gscbps.2019.9.3.0238 |
||||
| 94. Karas RA, Alexeree S, Elsayed H, Attia YA. Assessment of wound healing activity in diabetic mice treated with a novel therapeutic combination of selenium nanoparticles and platelets rich plasma. Sci Rep 2024; 14: 5346. doi:10.1038/s41598-024-54064-2 https://doi.org/10.1038/s41598-024-54064-2 PMid:38438431 PMCid:PMC10912747 |
||||
| 95. Luque Contreras D, Vargas Robles H, Romo E, Rios A, Escalante B. The role of nitric oxide in the post-ischemic revascularization process. Pharm Ther 2006; 112: 553-63. doi:10.1016/j.pharmthera.2006.05.003 https://doi.org/10.1016/j.pharmthera.2006.05.003 PMid:16950515 |
||||
| 96. Lessiani G, Vazzana N, Cuccurullo C, Di Michele D, Laurora G, Sgrò G, et al. Inflammation, oxidative stress and platelet activation in aspirin-treated critical limb ischaemia: Beneficial effects of iloprost. Thromb Haemost 2011; 105: 321-8. doi:10.1160/TH10-07-0499 https://doi.org/10.1160/TH10-07-0499 PMid:21103664 |
||||
| 97. Morita M, Hayashi T, Ochiai M, Maeda M, Yamaguchi T, Ina K, et al. Oral supplementation with a combination of l-citrulline and l-arginine rapidly increases plasma l-arginine concentration and enhances no bioavailability. Biochem Biophys Res Comm 2014; 454: 53-57. doi:10.1016/j.bbrc.2014.10.029 https://doi.org/10.1016/j.bbrc.2014.10.029 PMid:25445598 |
||||
| 98. Gorabi AM, Kiaie N, Hajighasemi S, Banach M, Penson PE, Jamialahmadi T, et al. Statin-induced nitric oxide signaling: Mechanisms and therapeutic implications. J Clin Med 2019 https://doi.org/10.3390/jcm8122051 PMid:31766595 PMCid:PMC6947613 |
||||
| 8: 2051. doi:10.3390/jcm8122051 https://doi.org/10.3390/jcm8122051 PMid:31766595 PMCid:PMC6947613 |
||||
| 99. Falconer D, Papageorgiou N, Salem K, Lim WY, Katsargyris A, Avgerinos E, et al. Nitric oxide donors for peripheral artery disease. Curr Opin Pharmacol 2018; 39: 77-85. doi:10.1016/j.coph.2018.02.009 https://doi.org/10.1016/j.coph.2018.02.009 PMid:29587164 |
||||
| 100. Wijaya A, Wang Y, Tang D, Zhong Y, Liu B, Yan M, et al, A study of lovastatin and l-arginine co-loaded PLGA nanomedicine for enhancing nitric oxide production and ENOS expression. J Mater Chem B 2022; 10: 607-24. doi:10.1039/D1TB01455B https://doi.org/10.1039/D1TB01455B PMid:34994373 |
||||
| 101. Ma X, Cheng Y, Jian H, Feng Y, Chang Y, Zheng R, et al. Hollow, rough, and nitric oxide-releasing cerium oxide nanoparticles for promoting multiple stages of wound healing. Adv Health Mater 2019; 8: 1900256. doi:10.1002/adhm.201900256 https://doi.org/10.1002/adhm.201900256 PMid:31290270 |
||||
| 102. Hou J, Pan Y, Zhu D, Fan Y, Feng G, Wei Y, et al.Targeted delivery of nitric oxide via a 'bump-and-hole'-based enzyme-prodrug pair. Nature Chem Biol 2019; 15: 151-60. doi:10.1038/s41589-018-0190-5 https://doi.org/10.1038/s41589-018-0190-5 PMid:30598545 PMCid:PMC10411945 |
||||
| 103. Madaric J, Valachovicova M, Paulis L, Pribojova J, Mateova R, Sebekova K, et al. Improvement in asymmetric dimethylarginine and oxidative stress in patients with limb salvage after autologous mononuclear stem cell application for critical limb ischemia. Stem Cell Res Ther 2017; 8: 165. doi:10.1186/s13287-017-0622-2 https://doi.org/10.1186/s13287-017-0622-2 PMid:28697789 PMCid:PMC5506609 |
||||
| 104. Zhang J, Qi H, Wang H, Hu P, Ou L, Guo, S, et al. Engineering of vascular grafts with genetically modified bone marrow mesenchymal stem cells on poly (propylene carbonate) graft. Artif Organs 2006; 30: 898-905. doi:10.1111/j.1525-1594.2006.00322.x https://doi.org/10.1111/j.1525-1594.2006.00322.x PMid:17181830 |
||||
| 105. Botusan IR, Sunkari VG, Savu O, Catrina AI, Grünler J, Lindberg S, et al. Stabilization of hif-1α is critical to improve wound healing in diabetic mice. Proc Nal Acad Sci 2008; 105): 19426-31. doi:10.1073/pnas.0805230105 https://doi.org/10.1073/pnas.0805230105 PMid:19057015 PMCid:PMC2614777 |
||||
| 106. Vincent KA, Shyu K-G, Luo Y, Magner M, Tio RA, Jiang C, et al. Angiogenesis is induced in a rabbit model of hindlimb ischemia by naked DNA encoding an HIF-1α/VP16 hybrid transcription factor. Circulation 2000; 102: 2255-61. doi:10.1161/01.CIR.102.18.2255 https://doi.org/10.1161/01.CIR.102.18.2255 PMid:11056102 |
||||
| 107. Rajagopalan S, Olin J, Deitcher S, Pieczek A, Laird J, Grossman PM, et al. Use of a constitutively active hypoxia-inducible factor-1α transgene as a therapeutic strategy in no-option critical limb ischemia patients. Circulation 2007; 115: 1234-43. doi:10.1161/CIRCULATIONAHA.106.607994 https://doi.org/10.1161/CIRCULATIONAHA.106.607994 PMid:17309918 |
||||
| 108. Sarkar K, Fox-Talbot K, Steenbergen C, Bosch-Marcé M, Semenza GL. Adenoviral transfer of hif-1α enhances vascular responses to critical limb ischemia in diabetic mice. Proc Nat Acad Sci 2009; 106: 18769-74. doi:10.1073/pnas.0910561106 https://doi.org/10.1073/pnas.0910561106 PMid:19841279 PMCid:PMC2774037 |
||||
| 109. Pei X, Kim H, Lee M, Wang N, Shin J, Lee S, et al. Local delivery of cardiac stem cells overexpressing HIF-1α promotes angiogenesis and muscular tissue repair in a hind limb ischemia model. J Contr Rel 2020; 322: 610-21. doi:10.1016/j.jconrel.2020.03.017 https://doi.org/10.1016/j.jconrel.2020.03.017 PMid:32194175 |
||||
Copyright

This work is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License.
AUTHOR'S CORNER
